2. Introduction
2.1. What is EDGE?
EDGE is a highly adaptable bioinformatics platform that allows laboratories to quickly analyze and interpret genomic sequence data. The bioinformatics platform allows users to address a wide range of use cases including assay validation and the characterization of novel biological threats, clinical samples, and complex environmental samples. EDGE is designed to:
Align to real world use cases
Make use of open source (free) software tools
Run analyses on small, relatively inexpensive hardware
Provide remote assistance from bioinformatics specialists
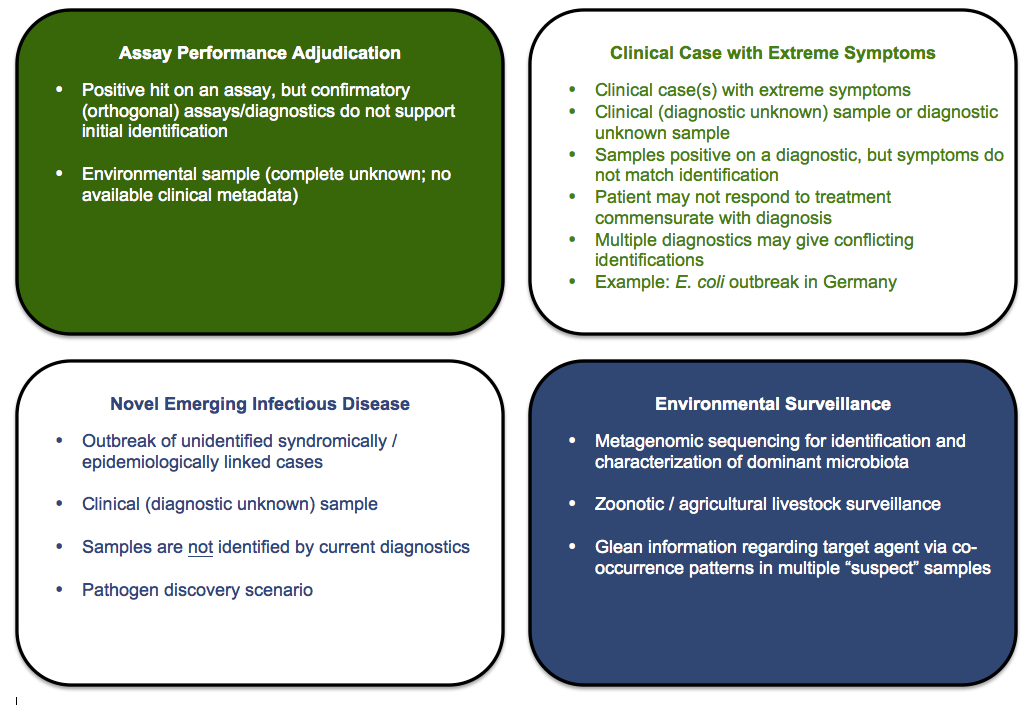
Four common Use Cases guided initial EDGE Bioinformatic Software development.
2.2. Why create EDGE?
EDGE bioinformatics was developed to help biologists process Next Generation Sequencing data (in the form of raw FASTQ files), even if they have little to no bioinformatics expertise. EDGE is a highly integrated and interactive web-based platform that is capable of running many of the standard analyses that biologists require for viral, bacterial/archaeal, and metagenomic samples. EDGE provides the following analytical workflows: quality trimming and host removal, assembly and annotation, comparisons against known references, taxonomy classification of reads and contigs, whole genome SNP-based phylogenetic analysis, and PCR analysis. EDGE provides an intuitive web-based interface for user input, allows users to visualize and interact with selected results (e.g. JBrowse genome browser), and generates a final detailed PDF report. Results in the form of tables, text files, graphic files, and PDFs can be downloaded. A user management system allows tracking of an individual’s EDGE runs, along with the ability to share, post publicly, delete, or archive their results.
While the design of EDGE was intentionally done to be as simple as possible for the user, there is still no single ‘tool’ or algorithm that fits all use-cases in the bioinformatics field. Our intent is to provide a detailed panoramic view of your sample from various analytical standpoints, but users are encouraged to have some insight into how each tool or workflow functions, and how the results should best be interpreted.